18
Historisch gesehen, wurden zunächst
Methoden aus der theoretischen Physik,
die den Festkörper als ein Kontinuum
beschreiben (die Methode der effektiven
Masse, oder K.p-Methode), herangezogen
und auf kleine, nanoskopische Systeme
angewandt. Obwohl formal dabei keine
Probleme entstehen, werden die Ergebnisse
unzuverlässig, sobald atomare Effekte rele-
vant werden, was zwangsläufig bei kleiner
werdenden Strukturen passiert.
In den letzten etwa zehn Jahren haben
sich die
ab
-
initio
-Methoden, bei denen die
Nanostruktur nicht als effektives Medium
einer gewissen Größe, sondern als eine
Anordnung verschiedener Atome beschrie-
ben wird, rasant entwickelt. Dabei unter-
scheiden wir drei verschiedene Ansätze.
Zum einen Methoden, die auf Greenschen
Funktionen und dem Bethe-Salpeter Ansatz
basieren, zum anderen Methoden, die auf
die Zeitabhängige Dichtefunktionaltheorie
zurückgreifen, und letztlich Methoden,
die den quantenchemischen Ansatz des
Quasiteilchen Konzepts verfolgen. Unser
AK entwickelt und verwendet den letzten
Ansatz. Abbildung 3 zeigt einen kolloi-
dalen Halbleiternanopartikel aus Cadmi-
umselenid. Dabei werden die Atome als
weiße und grüne Kugeln gezeigt und eine
ausgewählte Wellenfunktion als rote und
blaue Flächen dargestellt. Für solche Struk-
turen haben wir Methoden entwickelt,
die es erlauben, die Emissionswellenlänge
hochpräzise auszurechnen. Insbesondere
können wir die sogenannte Feinstruktur-
aufspaltung des Exzitons berechnen, die
energetisch imMikroelektronenvoltbereich
liegt, und die z. B. in der Quantenoptik von
großem Interesse ist.
Dynamik von angeregten Zuständen
Der oben beschriebene Ansatz berücksich-
tigt jedoch nicht das dynamische Verhal-
ten der untersuchten Systeme. Unsere
Aussagen über die Zerfallsraten entneh-
men wir lediglich einer Wahrscheinlich-
keitsrechnung. Es gibt aber Situationen,
bei denen diese Dynamik bedeutend zum
Vorschein kommt und die Zeitabhängigkeit
explizit berechnet werden muss. Dazu
gehören Diffusionsprozesse in Flüssigkei-
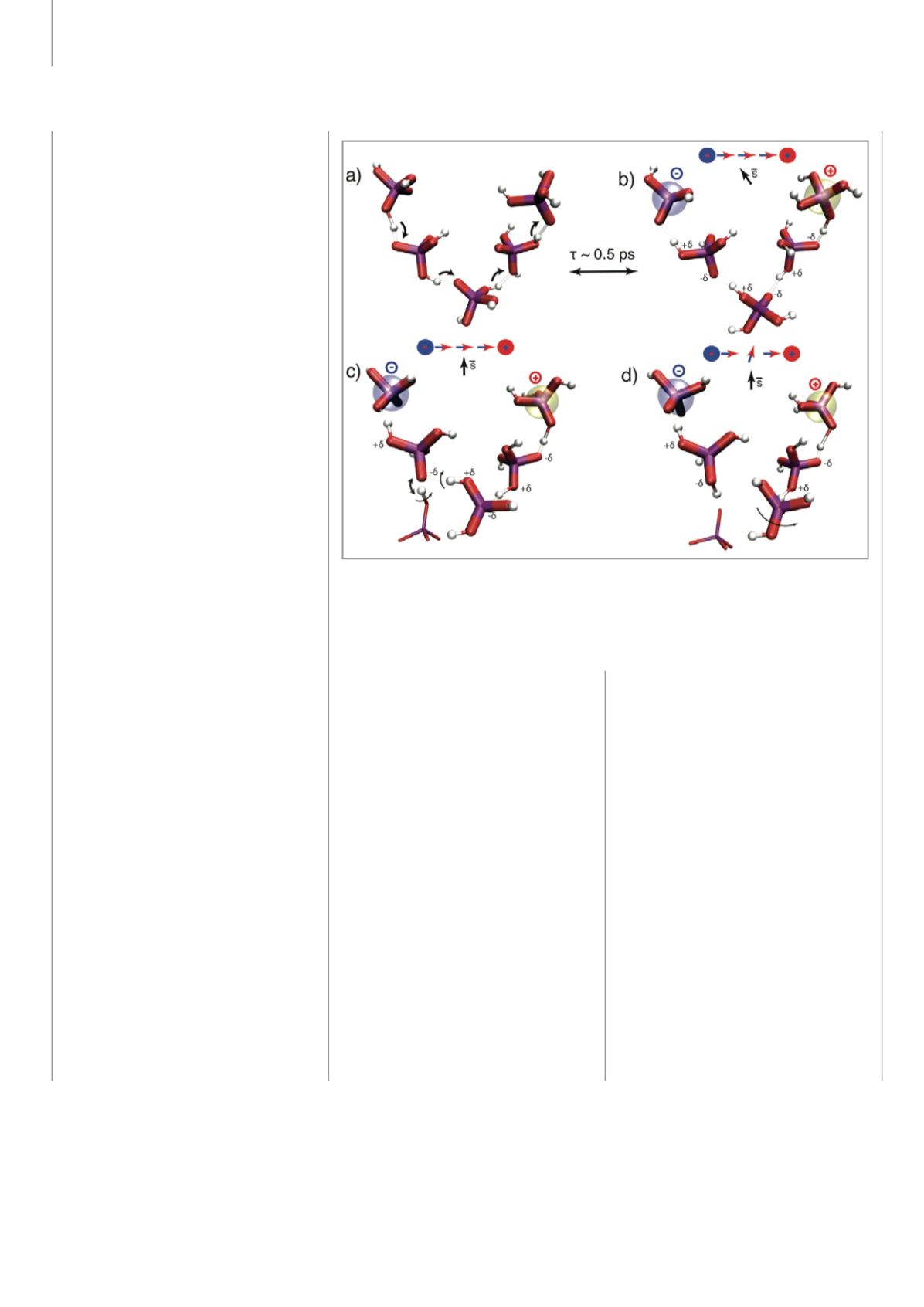
ten, wie z.B. die Migration von Protonen
in Phosphorsäure (siehe Abbildung 4). Die
Berechnung der atomaren Bewegung bei
einer eingestellten Temperatur von 400
Kelvin für etwa 50 Picosekunden erlaubt
es, den atomaren Diffusionsmechanismus
aufzuzeigen und die Diffusionskonstante
zu berechnen. Dabei handelt es sich bei den
Berechnungen um die Zeitabhängigkeit der
Entwicklung des Grundzustandes, bei dem
die Elektronen mühelos der Bewegung der
Kerne folgen. Diese Simulationen sind sehr
aufwendig, können aber mittels Standard-
methoden der
ab
-
initio
-Molekulardynamik
durchgeführt werden.
Wenn Nanostrukturen, wie in Hamburg
am CUI, durch intensive und kohärente
Lichtimpulse in einen angeregten Zustand
gebracht werden, relaxiert dieser nach
einer systemspezifischen Zeit wieder in den
Grundzustand. Die existierenden theore-
tischen Ansätze, um solche Prozesse zu
beschreiben, beruhen auf verschiedenen
Näherungen, die unter den neuartigen
experimentellen Bedingungen nicht mehr
gerechtfertigt und tragbar sind. Das Ziel
des Arbeitskreises ist die Erarbeitung von
analytischen Ansätzen und numerischen
Methoden, um die zeitliche Entwicklung
von angeregten Zuständen für Systeme
mit großer Atomanzahl zu berechnen.
Die Genauigkeit unserer Berechnungen
wird einerseits mit quantenchemischen
Verfahren, die zuverlässige Ergebnisse für
sehr kleine Modellsysteme liefern und
andererseits in direkter Zusammenarbeit
mit führenden experimentellen Gruppen
in Hamburg mit experimentellen Daten
verglichen.
Abbildung 4: Mikroskopischer Mechanismus des Protonen-Transports in Phosphorsäure aus
ab-initio-Molekulardynamik-Simulationen. Man beobachtet starke, polarisierbareWasser-
stoffbrücken und eine gekoppelte Protonenbewegung sowie eine ausgeprägte protonische
dielektrische Antwort des Mediums. Dieses Zusammenspiel erlaubt die Bildung ausgedehnter,
polarisierter Wasserstoffbrücken gebundener (Grotthuss-) Ketten.


